
Index
VaRank introduction
VaRank requirements
Input data
Output data
VaRank initial filtering
VaRank barcode
VaRank scoring
VaRank introduction
VaRank is a simple and powerful tool designed for variant ranking from next generation sequencing data.
It provides a comprehensive workflow for annotating and ranking SNVs and indels.
If you are interested by Structural Variation (SV) ANNOTATION and RANKING, please go to the AnnotSV software homepage.
Five modules create the strength of this workflow:
- Variant call quality summary (total and variant depth of coverage, phred like information), to filter out false positive calls
- Alamut Batch or SnpEff variant annotations, to integrate genetic and predictive information (functional impact, putative effects in the protein coding regions, population frequency...) from different sources, using HGVS nomenclature
- Barcode representing the presence/absence of variants (with homozygote/heterozygote status), to search for recurrence between families or group of individuals
- Phenotype-driven analysis, to score genes overlapped with a SNV/indel on biological relevance to the individual phenotype
- Prioritization score, to rank variants according to their predicted pathogenic status.
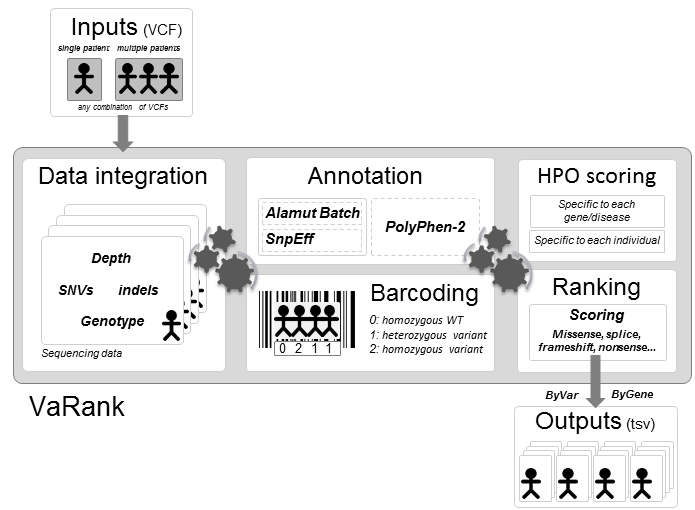
VaRank results aims at reducing the daily work of clinical geneticists and molecular biologists and will help to accelerate the progress in identifying disease causing variants.
VaRank requirements
a- You will need VaRank sources.
The Source code is available here under the GNU GPL licence.
b- VaRank can run on any architecture with a standard Tcl/Tk installation.
You can freely download it here for any architecture (e.g. AIX, Linux, Mac OS X, Solaris and Windows).
c- VaRank relies on 2 possible annotation engines to extract most of the data and offers the ability to score each variant:
Alamut Batch (Interactive Biosoftware). You can request a free, 30-day trial of Alamut Batch here.
SnpEff (http://snpeff.sourceforge.net).
Optional:
d- PolyPhen-2 provides prediction of functional effects of human SNPs. Depending on the annotation engine PPH2 either needs to be installed separately (Alamut Batch) or is already integrated (SnpEff). Nevertheless one can still have SnpEff installed and a local installation of PPH2.
You can freely download it
here
Input data
VaRank supports the commonly used VCF (Variant Call Format) input format for variants analysis that allows the program to be easily integrated into NGS bioinformatics analysis pipelines.
Output data
VaRank provides 4 tsv output files (TAB separated values files) divided into 2 categories:
-
Files named with “ByVar” contains variations sorted from the most to the least pathogenic (according to the VaRank score)
-
Files named with “ByGene” contains variations classified by gene (“ByGene”) where the list is sorted using the gene as a proxy to the score.
Each gene is scored according to most pathogenic variant (homozygous) or the first two most pathogenic variants.
In order to make sure that no variants are missed all gene variations are reported also below the variant(s) used to score the gene.
This file is more suitable when dealing with a recessive mode of inheritance.
A part from these 2 categories, each file is also available in 2 versions:
The description of the VaRank annotation columns is available in section 7 (“ANNOTATION COLUMNS”) of the README.VaRank_*.pdf.
VaRank initial filtering
The default filters remove variants:
-
with a total depth of coverage <= 10x
-
with a supporting reads count <= 10x
-
with a percent of supporting reads <= 15%
-
with validated annotation in the dbSNP database
(i.e. at least with 2 evidences) that are not pathogenic (from the ClinicalSignificance field in dbSNP)
-
with an allele frequency > 1% (extracted from the dbSNP, 1000Genomes, gnomad...)
VaRank barcode
VaRank introduces a barcode that allows a quick overview of the presence/absence status of each variant and their zygosity status within the analyzed individuals.
Together with the barcode, simple counts on the individuals (homozygous, heterozygous and total allelic counts) are also added and can easily be used to further filter variants not yet reported in dbSNP but present in the user’s cohort.
The combination of barcode and counts is an extremely powerful filtering strategy.

A. The barcode represents the SNV’s zygosity status in an ordered list of samples.
Samples homozygotes for the reference allele are represented using “0”, heterozygous variants are represented using “1” and homozygous variants are represented with “2”.
B. Selected annotations from the VaRank output representing 3 SNVs from a single patient.
The barcode gives an overview of the presence/absence for one SNV in all other patients analyzed.
Together with this, the total counts of alleles are given in the last 4 columns.
In a cohort of 32 samples, the variant in BBS2 is present in 31/32 samples at the homozygous state as one can see from the barcode or the relevant counts.
The variant in ALMS1 (disease causing mutation) is present once at the homozygous state.
C: The barcode can be specifically ordered and used in family analysis such as trio exome sequencing.
On the left, homozygous mutations in a consanguineous family could be highlighted by the “121” barcode indicating homozygous variants (“2”) in the proband inherited from heterozygous parents (“1”).
On the right denovo variants in the proband could be highlighted with the proposed barcode “010”.
HPO scoring
To score genes overlapped with a SNV/indel on biological relevance to the individual phenotype, VaRank rely on Exomiser (Smedley et al., 2015) and HPO (Köhler et al., 2019).
For a given phenotype, a HPO-based score corresponding to a damaging probability is provided for each gene overlapped with an SNV/indel so that:
Genes previously associated with disease can be highlighted easily
Genes not previously associated with disease can be highlighted
Genes associated with diseases that have little or no similarity to the observed phenotypes can be removed along
HPO:
VaRank uses the Human Phenotype Ontology (version reported in the VaRank output). Find out more at the Human Phenotype Ontology website.
Please cite the 2 following articles if you use these phenotype data in your work:
Next-generation diagnostics and disease-gene discovery with the Exomiser. Smedley D., et al, Nature Protocols (2015) doi:10.1038/nprot.2015.124
Expansion of the Human Phenotype Ontology (HPO) knowledge base and resources. Köhler S., et al, Nucleic Acids Research (2019) doi:10.1093/nar/gky1105
VaRank scoring
The description of the VaRank scores is detailed in section 5 (“SCORING”) of the README.VaRank_*.pdf.
If you have any problem or question, please, feel free to contact us at jeanmuller@unistra.fr or veronique.geoffroy@inserm.fr