Subsections
6.1 The Ordalie database scheme
The core of Ordalie is build around an in-memory SQLite database [3] which scheme is given in figure XXX.
Ordalie takes advantage of this underlaying database to store snapshots of alignments and their associated features.
The "ordalie" table contains settings parameters saved at exit allowing the user to find the same state when launching Ordalie again.
The "seqinfo" table contains sequence information that are not linked to amino acids positions (length, molecular weight, isoelectric point, ...)
The "seqfeat" table is used to store features data mapped onto the residue sequence.
Upon loading of a new alignment file, Ordalie creates a first snapshot as being a read-only copy of this alignment stored as the "original alignment" in the snapshot table. This table contains all snapshots created so far along with their name and description.
The "seqali" table records the amino acid sequences as they appear in the snapshots.
A link table "ln_snapshot_seqali" binds a given set of sequences to a given snapshot.
Accordingly, the "featali" table stores features attached to aligned sequences in a given snapshot. A link table "ln_seqali_featali" couple these two tables.
The "clustering" and "cluster" tables define a given clustering attached to a snapshot with its name, the method and residue zones used to compute it, and the resulting clusters with their names respectively.
The set of sequences defining a given cluster is available through the "ln_seqali_cluster" link table.
The "colmeasure" and "colscore" tables correspond to conservation computations (column measurements) with their name and used method, and the conservation groups with name, value for each column of the group respectively.
The conservation score for a given cluster is available through the link table "ln_cluster_colscore".
Finally, the "annotation" table contains all information relative to annotation the user adds to a given snapshot.
The Ordalie (. ord file extension) consists in a database dump.
6.2 The FindPatterns syntax
The search pattern can include any legal sequence character, and also include several non-sequence characters, which are used to specify 'OR' matching, 'NOT' matching, 'begin' and 'end' constraints, and repeat counts. For instance, the pattern GASTE(X){20,30}FTG means searching GASTE, followed by 20 to 30 of any amino acid, followed by FTG. Following is an explanation of the syntax for pattern specification.
Parentheses () enclose one or more symbols that can be repeated a certain number of times. Braces {} enclose numbers indicating how many times the symbols within the preceding parentheses must be found.
Sometimes, it is possible to leave out part of an expression. If braces appear without preceding parentheses, the numbers in the braces define the number of repeats for the immediately preceding symbol. One or both of the numbers within the braces may be missing. For instance, both the pattern GASG{2,}F and the pattern GASG{2}F mean GAS, followed by G repeated from 2 to 350,000 times, followed by F; the pattern GASG{}F means GAS, followed by G repeated from 0 to 350,000 times, followed by F; the pattern GAS(TE){,2}F means GAS, followed by TE repeated from 0 to 2 times, followed by F; the pattern GAS(TE){2,2}F means GAS, followed by TE repeated exactly 2 times, followed by F (If the pattern in the parentheses is an OR expression (see below), it cannot be repeated more than 2,000 times).
Specifying several symbol choices can be easily done by enclosing the different choices between parentheses and separating the choices with commas. For instance, RGF(Q,A)S means RGF followed by either Q or A followed by S. The length of each choice need not be the same, and there can be up to 31 different choices within each set of parentheses. The pattern GAT(TG,T,G){1,4}A means GAT followed by any combination of TG, T, or G from 1 to 4 times followed by A. The sequence GATTGGA matches this pattern. There can be several parentheses in a pattern, but parentheses cannot be nested.
The pattern GC~CAT means GC, followed by any symbol except C, followed by AT. The pattern GC~(A,T)CC means GC, followed by any symbol except A or T, followed by CC.
The pattern <GACCAT can only be found if it occurs at the beginning of the sequence range being searched. Likewise, the pattern GACCAT> would only be found if it occurs at the end of the sequence range.
6.3 The Vector Norm scoring method
This method is based on a vectorial representation of the 20 amino acids. This representation can be the same as the one used in the VRP representation, or can be for example, a volume/polarity couple. The score for a given column 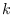 can be computed then by:
can be computed then by:
S(k) = nc/nt * |sum_i=1^nV|/sum_1=1^n|Vi|
where nc is the number of residues in the column, nt is the total number of sequences.
This function is bounded by 0. and N, where N is the number of sequences in the alignment.
6.4 The Feature File Format
It is possible to import features into Ordalie through a features file. It is also possible to add items to an existing feature, or to create completely new ones.
The feature file format looks like:
# This is an example of a feature file format
# A line starting by \# is a comment line that can be inserted everywhere
# Declare the feature
FEATURE MyFeat? PROPAGATE? ?all|group?
#
# Structure of the feature item:
# seq. name; coord. system; start; stop; color; score; note
Q65P3D; LOCAL; 23; 57; red; 0. 0; first item
Q65P3D; GLOBAL; 212; 345; blue; 0. 0; second one
FLK14Q; local; 123; 234; red; 0. 0; one more
# Then go to an other feature
FEATURE STRUCT
P12345; global; 2112; 2541; green; 0. 0; add one
To add some items to an existing feature, the feature name should be exactly identical to the one already present in the alignment as feature names are case-sensitive.
6.5 The superposition algorithm
Given two sets of atomic coordinates A and B, the following algorithm will try to minimize the RMS (Root Mean Squared) distance between A and B by moving B onto A. The algorithm can be separated in the following steps:
- Compute the centre of mass (CDM) of the two sets and translate B atoms by the vector joining B CDM to A,
- Compute the 3 main inertia axis for the two sets, which correspond to the three eigen vectors of the Eigen decomposition of the whole atomic coordinates set,
- By turn, minimize the weighted RMS by rotating around the Eulerian angle associated to the current axis.
- Stops when converged, and issue superposition information.
The output of this algorithm provides along with the RMS, the orientation matrix, translation vector, and rotations between the two molecules in different forms.
6.6 The command line options
|
Option |
Values |
Description |
|---|
|
-convert |
<TFA|MSF|XML|ORD> |
Converts the alignment into the format indicated by -convert. The converted output file name will have the form <alignemnt file>. <format> |
|
-precompute |
<0|1> |
: precompute clustering and conservation for each PFAM domain |
|
-threshold |
<x> |
Set conservation threshold level. <x> should be set between 51 and 100 |
|
-batch |
<0|1> |
Run Ordalie without windows and exit when finished |